Case study · Bioinformatics
NovoRank
A machine learning post-processing tool that re-ranks de novo peptide sequencing results by analysing similar spectra together — improving identification accuracy across existing tools, with no change to the tools themselves.
- Role
- Graduate Research Assistant · M.S. thesis, Hanyang University
- Timeline
- 2020 – 2022 · Published in JPR, 2025
- Stack
- Python TensorFlow scikit-learn
- Links
- GitHub Paper (PDF) Dataset
01. Problem
De novo sequencing identifies peptide sequences directly from mass-spectrometry data, without relying on a protein database. That makes it essential for finding novel peptides — for example, neoantigen discovery in immunotherapy.
But conventional de novo tools score each spectrum in isolation and lean on imperfect scoring functions, so they frequently rank the wrong peptide first — especially near the peptide ends, where key fragment ions are often missing and the amino-acid order turns ambiguous. The information sitting in other, similar spectra — which likely come from the same peptide — goes unused.
02. Approach
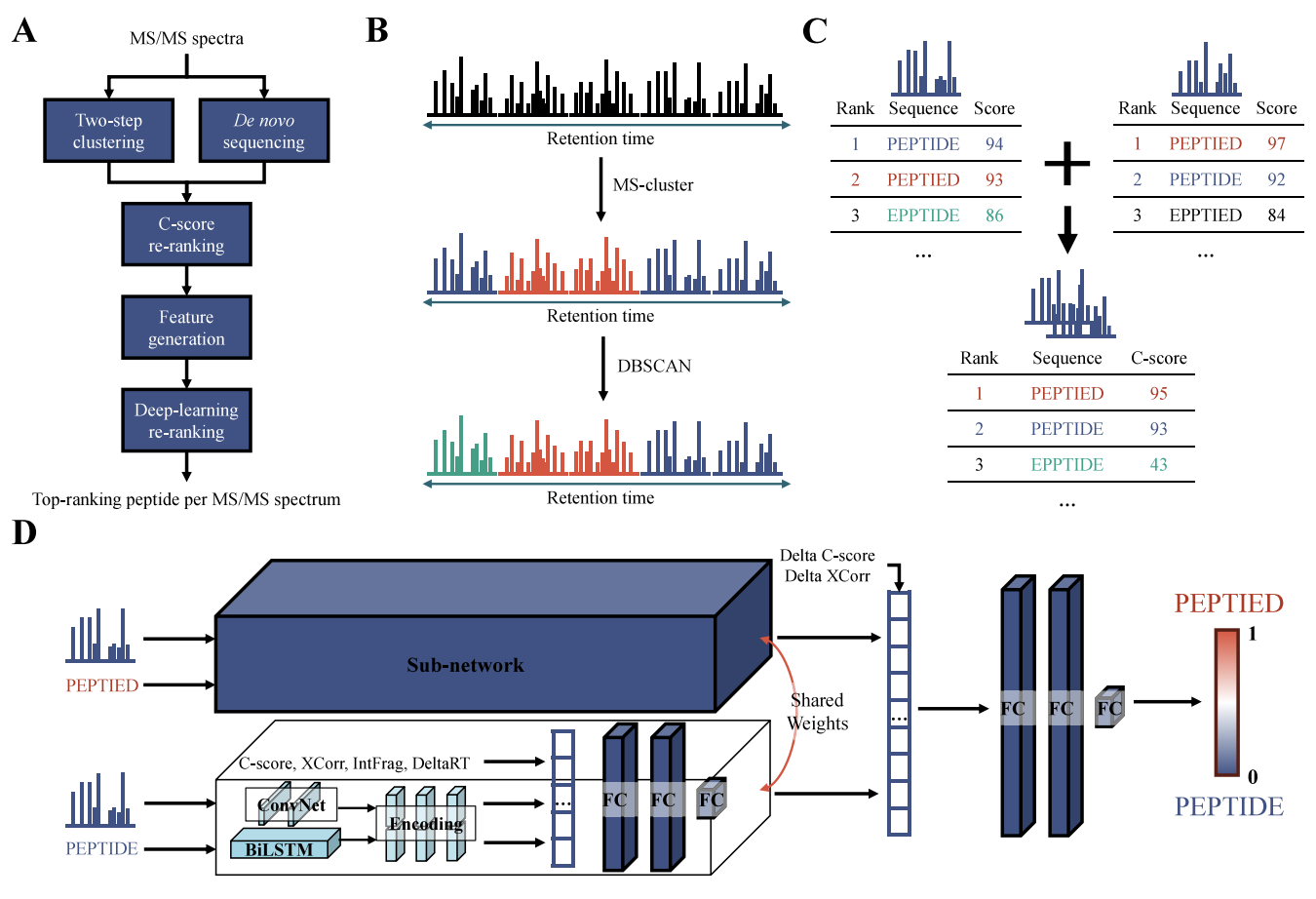
NovoRank re-ranks candidate peptides by analysing similar spectra together, assuming they originate from the same peptide species. Four pieces make that work:
-
Two-step clustering
Spectral Clustering groups similar spectra first, then DBSCAN refines each group, so spectra from the same peptide land together more precisely.
-
C-score
A cluster-aware score that normalises the summed de novo scores of a peptide by cluster size. Peptides that score well and recur across a cluster rank higher, while clusters of different sizes stay comparable.
-
A deep learning re-ranker
A siamese model with shared weights compares the top two candidates and picks the better one (learning-to-rank). It fuses three views of each candidate: the spectrum through a sub-network, the peptide sequence through a ConvNet + BiLSTM, and six tabular features (C-score, XCorr, fragment-ion match, ΔRT, …) through dense layers.
-
Tool-agnostic by design
NovoRank sits downstream of existing de novo tools, so it improves their precision and recall without modifying the tools themselves.
The quiet hard part was the clustering and the C-score. They look simple, but landing on features that powerful took real proteomics understanding and a long stretch of trial and error — I'd chased several new spectrum-to-peptide matching scores that went nowhere before these worked. As first author I performed the proteomic data analysis and developed and implemented the full tool.
03. Results
Evaluated across three de novo sequencing tools and three datasets:
+4.6%
average precision
+4.5%
average recall
+3.8% / +3.6%
precision / recall on Casanovo (SOTA)
- Correct peptide identifications rose by 0.37–0.61% (Casanovo), 7.52–18.80% (pNovo3), and 3.06–4.24% (PEAKS). Some peptides were lost, but more new ones were gained.
- Tools evaluated: PEAKS (commercial, algorithmic), pNovo3 (algorithm + ML), and Casanovo (transformer-based, state of the art).
04. Limitations & what's next
- The gains are real but modest, and for the strongest tool (Casanovo) the improvement is smaller at the peptide level than at the PSM level.
- NovoRank re-ranks at the PSM level, but the real evidence lives at the cluster level — multiple spectra from the same peptide. A cluster-level identification score, rather than per-PSM, is the natural next step.
05. Reference
Jangho Seo, Seunghyuk Choi, Eunok Paek. NovoRank: Refinement for De Novo Peptide Sequencing Based on Spectral Clustering and Deep Learning. J. Proteome Res. 2025, 24, 2, 903–910.
Read paper →